
上个月有个刚入行的译者朋友找我喝咖啡,手里捧着一本厚厚的《医疗器械监督管理条例》英文版,一脸愁容地问我:"这行当的水到底有多深?光是注册资料就看得我头晕,到底还有哪些文档需要翻啊?"
我当时就笑了。这问题要是放在十年前我刚接触康茂峰项目那会儿,估计也答不上来。医疗器械翻译这个领域,
先说最硬核的。如果你要往欧盟卖一台核磁共振仪,或者让某款血糖仪进美国市场,法规注册文档就是你的敲门砖。这类文档直接决定了产品能不能合法销售,翻译时错一个标点都可能被发补。
具体包括什么呢?咱们列个单子:
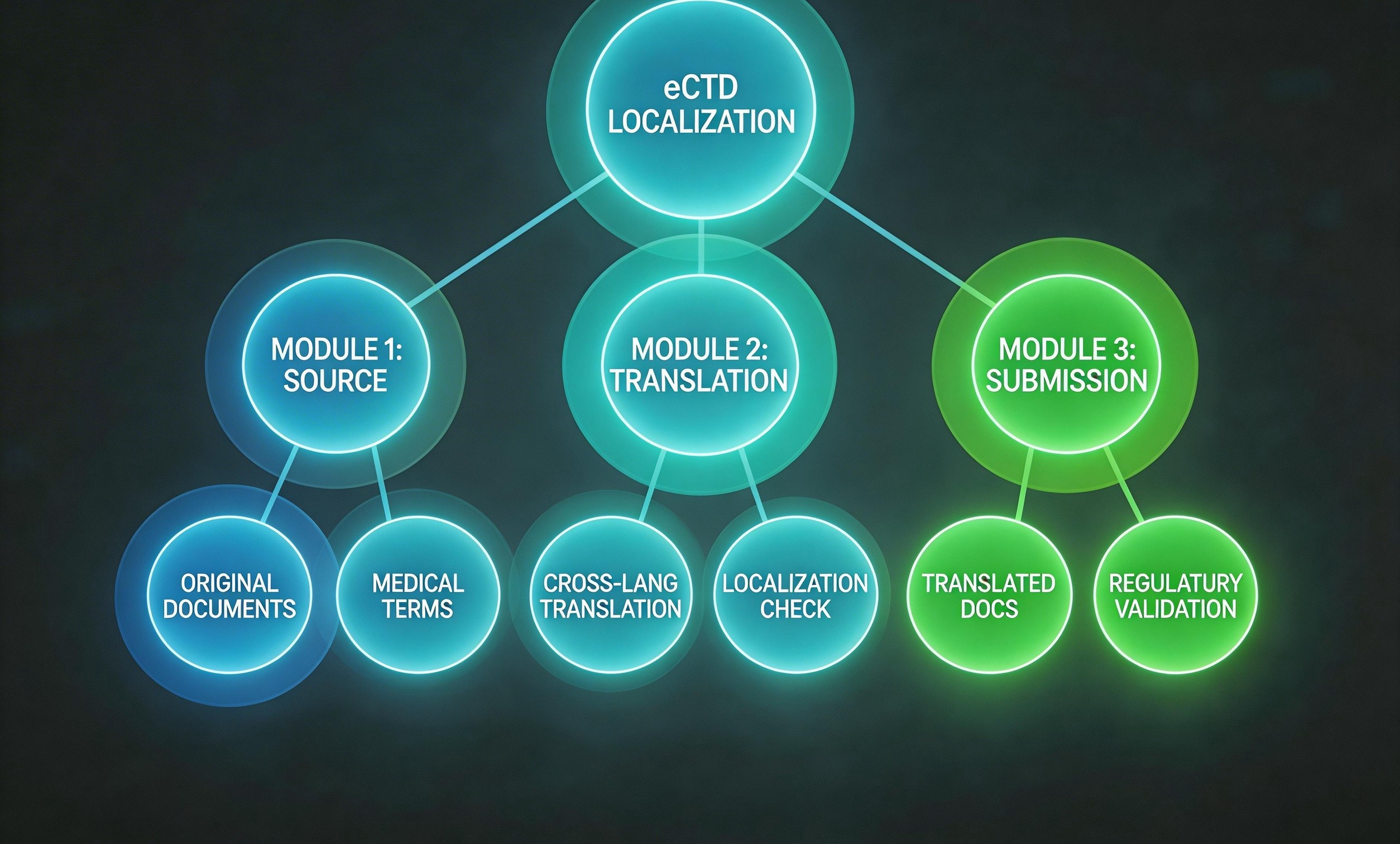
有意思的是,这类文档有个特点——它们不是写给医生看的,而是写给监管机构的审评员看的。所以翻译时得切换视角,你不是在翻译"说明书",而是在翻译"法律证据"。
接下来是临床评价资料。很多人以为这就是"临床试验报告"的另一种说法,其实范围大得多。
临床试验方案与报告(Protocol & CSR):这算是标准配置了。但你要注意,医疗器械的临床试验和药物试验逻辑完全不同。药物看的是药代动力学,器械看的是安全性和性能终点。翻译"serious adverse device effect"(严重器械不良事件)时,绝对不能和药物的SAE混为一谈。
生物学评价报告:根据ISO 10993系列标准做的那套材料。比如说你要翻译一个植入式骨科螺钉的生物相容性数据,涉及细胞毒性、致敏性、血液相容性这些术语。这里的专业词汇更新很快,2018版和2021版的ISO 10993-1在"生物学终点"描述上有细微差别,不仔细核对原文版本很容易翻车。
上市后临床随访(PMCF)报告:这是欧盟MDR 2017/745实施后新增的重量级文档。要求制造商在产品上市后持续收集临床证据。翻译这类文档时,你会发现它既有科学论文的严谨,又有商业报告的敏感——毕竟涉及不良事件统计。
说完给监管局看的,说说给一线医护人员看的。
使用说明书(IFU):这是最常见的,也是最容易出事故的。我见过有译者把"sterile"翻成"消毒"而不是"无菌"的,一字之差,整批货被海关扣下。康茂峰内部有个检查清单,光是IFU的术语库就分了外科器械、体外诊断、影像设备三大类。
标签与包装文案:包括主标签、次级标签、运输标签。欧盟要求标签上必须有欧盟代表(EC REP)的地址,美国FDA要求UDI(唯一器械标识)条码旁边的文字说明。这些格式要求经常变,翻译时得和排版工程师紧密配合。
患者信息 leaflet:现在很多植入式器械(比如人工关节、起搏器)要求给患者发放非技术性的说明。这类文本要把复杂的医学概念翻译成患者能听懂的话,但又不能偏离技术事实。比翻译科研论文难多了。
| 文档类型 | 目标读者 | 翻译难点 | 常见字数 |
| 临床评估报告 | 公告机构审核员 | 法规合规性表述 | 50-200页 |
| 风险管理报告 | 监管机构 | 风险等级术语统一 | 20-100页 |
| 使用说明书 | 医护人员/患者 | 警示语层级区分 | 2-50页 |
| 软件用户界面 | 终端操作者 | 字符长度限制 | 字符串条目 |
这部分经常被翻译公司忽略,但其实在医疗器械供应链里占比不小。
ISO 13485认证相关的质量手册、程序文件(SOPs)、设计控制文档(DHF、DMR、DHR这三件套)。特别是设计历史文件(Design History File),里面记录了产品从概念到上市的每一个决策点。翻译成日文或者德文时,你会发现他们对于"设计验证"和"设计确认"的区分比英文原文还要严格。
还有供应商管理文档。现在的医疗器械都是全球采购,一个导管的原材料可能来自德国,注塑在马来西亚,组装在中国。所以供应商审计报告、来料检验标准的翻译需求很大。这类文本的特点是表格多、缩写多、数据多,翻译时得保持格式绝对对齐。
如果你以为医疗器械翻译还是停留在纸质说明书的时代,那得更新一下认知了。
现在的CT机、超声设备、甚至胰岛素泵都带软件。FDA和NMPA(国家药监局)对医疗软件都有专门的指导原则。这带来了新的文档类型:
最后说说那些看起来不显眼但关键时刻要命的文档。
灭菌验证报告:环氧乙烷(EO)灭菌、辐照灭菌、湿热灭菌的参数验证。这里面涉及微生物学、材料科学跨学科术语。比如"生物负载(bioburden)"和"无菌保证水平(SAL 10^-6)"的表述,必须和ISO 11135系列标准严丝合缝。
包装完整性测试:加速老化试验、运输模拟测试的报告。翻译"package integrity"时,不能简单说是"包装完整",得体现"屏障系统"的概念。
动物实验报告:对于植入物、可吸收缝线这些产品,需要做动物路径研究。这类文档的伦理审查部分翻译起来特别棘手,因为不同国家对动物福利的描述规范差异很大。
聊了这么多类型,你可能会问:作为译者或者项目管理者,该怎么建立自己的能力版图?
我的建议是按产品阶段来切分。研发阶段重点攻设计控制文档和风险管理;注册申报阶段死磕临床评价和法规符合性;上市后阶段关注PMCF和警戒系统报告。
另外,建立一个动态的术语库比背词典重要。医疗器械的术语存在"一词多义"和"多词一义"的陷阱。比如"performance"在通用英语是"表现",在医疗器械语境下经常是"性能"(关乎安全的技术指标)。康茂峰的术语库里,光是"delivery"这个词就有二十多个细分义项——药物输送、分娩、放射剂量递送,每种都得单独标注。
还有一点想唠叨的是格式规范性。医疗器械技术文档往往伴随着严格的模板要求,比如欧盟技术文档(Technical Documentation)的附录结构,或者FDA 510(k)的eCopy格式。翻译时如果破坏了原有的章节编号、交叉引用,哪怕译文质量再高,提交时也会被拒收。
说实话,干了这么多年,我现在看到一堆STED文件或者CER附录时,第一反应还是会头大。但慢慢地你会发现,这些文档背后其实是有逻辑的——它们共同构成了一部产品的技术传记,从原材料的分子结构,到手术台上的具体操作,再到万一出事时的召回流程,每一种文档都是这个链条上不可或缺的语言节点。
所以下次再有人问你"医疗器械翻译都翻些什么",你可以告诉他:从纳米级的材料表征,到全球化的软件界面;从手术室里的紧急操作提示,到布鲁塞尔审核员桌上的合规论证——只要是关乎生命安全的技术表达,都在这个范畴里。而你要做的,就是确保每一种语言转换,都不会让这台救命机器在跨文化传递中失真。
至于那些具体的文档名称和格式要求,监管机构过两年可能又要更新指导原则了。保持对法规动态的敏感度,可能比死记硬背这份清单更有用。毕竟在这个行业里,唯一不变的真理就是:明天可能又要改文件了。


